fwdpy
forward-time population genetic simulation in python
HomeReference manual
Google Group
Tracking mutation frequencies
%matplotlib inline
%pylab inline
import fwdpy as fp
import pandas as pd
import matplotlib
import matplotlib.pyplot as plt
import copy
/usr/local/lib/python2.7/dist-packages/matplotlib/font_manager.py:273: UserWarning: Matplotlib is building the font cache using fc-list. This may take a moment.
warnings.warn('Matplotlib is building the font cache using fc-list. This may take a moment.')
Populating the interactive namespace from numpy and matplotlib
Run a simulation
nregions = [fp.Region(0,1,1),fp.Region(2,3,1)]
sregions = [fp.ExpS(1,2,1,-0.1),fp.ExpS(1,2,0.01,0.001)]
rregions = [fp.Region(0,3,1)]
rng = fp.GSLrng(101)
popsizes = np.array([1000],dtype=np.uint32)
popsizes=np.tile(popsizes,10000)
#Initialize a vector with 1 population of size N = 1,000
pops=fp.SpopVec(1,1000)
#This sampler object will record selected mutation
#frequencies over time. A sampler gets the length
#of pops as a constructor argument because you
#need a different sampler object in memory for
#each population.
sampler=fp.FreqSampler(len(pops))
#Record mutation frequencies every generation
#The function evolve_regions sampler takes any
#of fwdpy's temporal samplers and applies them.
#For users familiar with C++, custom samplers will be written,
#and we plan to allow for custom samplers to be written primarily
#using Cython, but we are still experimenting with how best to do so.
rawTraj=fp.evolve_regions_sampler(rng,pops,sampler,
popsizes[0:],0.001,0.001,0.001,
nregions,sregions,rregions,
#The one means we sample every generation.
1)
rawTraj = [i for i in sampler]
#This example has only 1 set of trajectories, so let's make a variable for thet
#single replicate
traj=rawTraj[0]
print traj.head()
print traj.tail()
print traj.freq.max()
esize freq generation origin pos
0 -0.314966 0.0005 1 0 1.382760
1 -0.021193 0.0005 1 0 1.367676
2 -0.066601 0.0005 1 0 1.125086
3 -0.066601 0.0005 2 0 1.125086
4 -0.066601 0.0010 3 0 1.125086
esize freq generation origin pos
104420 -0.016016 0.0005 9999 9998 1.773315
104421 -0.155373 0.0005 9999 9998 1.912775
104422 -0.155373 0.0005 10000 9998 1.912775
104423 -0.042471 0.0005 10000 9999 1.738310
104424 -0.030944 0.0005 10000 9999 1.805271
1.0
Group mutation trajectories by position and effect size
Max mutation frequencies
mfreq = traj.groupby(['pos','esize']).max().reset_index()
#Print out info for all mutations that hit a frequency of 1 (e.g., fixed)
mfreq[mfreq['freq']==1]
| pos | esize | freq | generation | origin | |
|---|---|---|---|---|---|
| 2701 | 1.134096 | 0.001812 | 1.0 | 2612 | 43 |
The only fixation has an ‘esize’ $> 0$, which means that it was positively selected,
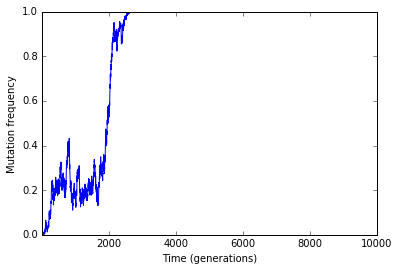
Frequency trajectory of fixations
#Get positions of mutations that hit q = 1
mpos=mfreq[mfreq['freq']==1]['pos']
#Frequency trajectories of fixations
fig = plt.figure()
ax = plt.subplot(111)
plt.xlabel("Time (generations)")
plt.ylabel("Mutation frequency")
ax.set_xlim(traj['generation'].min(),traj['generation'].max())
for i in mpos:
plt.plot(traj[traj['pos']==i]['generation'],traj[traj['pos']==i]['freq'])
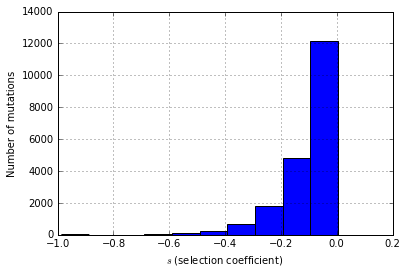
#Let's get histogram of effect sizes for all mutations that did not fix
fig = plt.figure()
ax = plt.subplot(111)
plt.xlabel(r'$s$ (selection coefficient)')
plt.ylabel("Number of mutations")
mfreq[mfreq['freq']<1.0]['esize'].hist()
<matplotlib.axes._subplots.AxesSubplot at 0x7f396f0f7090>