Molecular population genetic analysis.
More...
#include <Sequence/PolySNP.hpp>
Molecular population genetic analysis.
Example Use:
#include <iostream>
#include <vector>
int main
{
vector<Fasta> data;
Alignment::GetData(data,"popdata.fasta");
assert(Alignment::IsAlignment(data))
if (Alignment::Gapped(data))
{
Alignment::RemoveTerminalGaps(data);
}
cout <<
"Tajima's D is " << analyze->
TajimasD() << endl;
delete polytable;
delete analyze;
exit(1);
}
- Warning
- The routines to calculate nucleotide diversity, numbers of singletons, etc. all handle missing data (the
N character). However, all summary statistics involved in "tests of
neutrality" are, strictly speaking, undefined if missing data are present. The reason for this is because the denominators of the statistics are functions of the sample sizes, and no explicit formulae exist when the sample size varies from site to site (which is the case when there are missing data). In short, if you want to be rigorous, you can only really count up nucleotide diversity and a few other statistics if your data contain untyped SNPs. However, the routines present in libsequence will happily go and calculate the summary statistics for you, and it is up to you to be aware that you are writing a program that may give biased results. To date, the magnitude and direction of the bias remains unknown. Functions (and hence the statistics) that are affected have warnings in their documentation.
- Note
- As of libsequence 1.4.1, the routines in this class explicity check for gaps in the polymorphism table. This provides an additional safeguard for cases where Sequence::PolyTable objects are created and sites with gaps are left in.
- Deprecated:
- Will be removed in libsequence 2.0
- Examples:
- slidingWindow.cc, and slidingWindow2.cc.
Definition at line 86 of file PolySNP.hpp.
◆ PolySNP()
| Sequence::PolySNP::PolySNP |
( |
const Sequence::PolyTable * |
data, |
|
|
bool |
haveOutgroup = false, |
|
|
unsigned |
outgroup = 0, |
|
|
bool |
totMuts = true |
|
) |
| |
|
explicit |
- Parameters
-
| data | a valid object of type Sequence::PolyTable |
| haveOutgroup | true if an outgroup is present, false otherwise |
| outgroup | if haveOutgroup is true, outgroup is the index of that sequence in data |
| totMuts | if true (the default) use the total number of inferred mutations, otherwise use the total number of polymorphic sites in calculations |
- Note
- this constructor allocates a pointer to an implementation class, which automatically pre-processes the SNP data
Definition at line 171 of file PolySNP.cc.
◆ a_sub_n()
| double Sequence::PolySNP::a_sub_n |
( |
void |
| ) |
const |
|
protected |
![\[a_n=\sum_{i=1}^{i=n-1}\frac{1}{i}.\ \]](form_40.png)
This is the denominator of Watterson's Theta (see PolySNP::ThetaW)
- Warning
- statistic undefined if there are untyped SNPs
Definition at line 1504 of file PolySNP.cc.
◆ a_sub_n_plus1()
| double Sequence::PolySNP::a_sub_n_plus1 |
( |
void |
| ) |
const |
|
protected |
![\[a_{n+1}=\sum_{i=1}^{i=n}\frac{1}{i}\ \]](form_41.png)
- Warning
- statistic undefined if there are untyped SNPs
Definition at line 1520 of file PolySNP.cc.
◆ b_sub_n()
| double Sequence::PolySNP::b_sub_n |
( |
void |
| ) |
const |
|
protected |
![\[b_n=\sum_{i=1}^{i=n-1}\frac{1}{i^2}\ \]](form_42.png)
- Warning
- statistic undefined if there are untyped SNPs
Definition at line 1537 of file PolySNP.cc.
◆ b_sub_n_plus1()
| double Sequence::PolySNP::b_sub_n_plus1 |
( |
void |
| ) |
const |
|
protected |
![\[b_n=\sum_{i=1}^{i=n}\frac{1}{i^2}\ \]](form_43.png)
- Warning
- statistic undefined if there are untyped SNPs
- Author
- Joshua Shapiro
Definition at line 1552 of file PolySNP.cc.
◆ c_sub_n()
| double Sequence::PolySNP::c_sub_n |
( |
void |
| ) |
const |
|
protected |
![\[ c_n=\left\{\begin{array}{cl} 1 , & when\ n = 2 \\ \frac{2 \times (n \times a_n - 2 \times (n-1))}{(n-1) \times (n-2)}, & when\ n > 2 \\ \end{array}\right.\ \]](form_44.png)
- Warning
- statistic undefined if there are untyped SNPs
Definition at line 1568 of file PolySNP.cc.
◆ d_sub_n()
| double Sequence::PolySNP::d_sub_n |
( |
void |
| ) |
const |
|
protected |
![\[\ d_n=\frac{2}{n-1} \times (1.5 - \frac{2 \times a_{n+1}}{n-2} - \frac{1}{n})\ \]](form_45.png)
- Warning
- statistic undefined if there are untyped SNPs
Definition at line 1595 of file PolySNP.cc.
◆ DandVH()
| double Sequence::PolySNP::DandVH |
( |
void |
| ) |
const |
To check if two sequences are unique, Sequence::Comparisons::Different is used, which does not allow missing data to result in 2 sequences being considered different (as they would be if you simply used thestd::string comparison operators == or !=)
- Returns
- the haplotype diversity of the data.
Definition at line 1614 of file PolySNP.cc.
◆ DandVK()
| unsigned Sequence::PolySNP::DandVK |
( |
void |
| ) |
const |
To check if two sequences are unique, Sequence::Comparisons::Different is used, which does not allow missing data to result in 2 sequences being considered different (as they would be if you simply used the std::string comparison operators == or !=)
- Returns
- number of haplotypes in the sample
Definition at line 1631 of file PolySNP.cc.
◆ DepaulisVeuilleStatistics()
| void Sequence::PolySNP::DepaulisVeuilleStatistics |
( |
void |
| ) |
const |
|
protected |
Calculate the number of haplotypes in the sample, and haplotype diversity. Unlike Depaulis and Veuille's original paper, this routine uses an unbiased calculation of haplotype diversity (i.e. divide by n choose 2).
To check if two sequences are unique, Sequence::Comparisons::Different is used, which does not allow missing data to result in 2 sequences being considered different (as they would be if you simply used the std::string comparison operators == or !=)
Definition at line 1050 of file PolySNP.cc.
◆ Disequilibrium()
| std::vector< PairwiseLDstats > Sequence::PolySNP::Disequilibrium |
( |
const unsigned & |
mincount = 1, |
|
|
const double & |
max_marker_distance = std::numeric_limits<double>::max() |
|
) |
| const |
- Returns
- A vector of statistics related to LD and distance in the sample. An empty vector is returned if there are < 2 polymorphic sites in the sample. See the documentation for Recombination::Disequilibrium for a description of the return vector.
- Parameters
-
| mincount | a frequency filter. A polymorphism must be present at least mincount times in the data |
- Note
- For D and D', the 11 gamete is defined as follows: If no outgroup is present, it refers to the genotype of minor alleles at both sites. If there is an outgroup, it is based on the genotype of derived alleles at both sites.
Definition at line 1837 of file PolySNP.cc.
◆ Dnominator()
| double Sequence::PolySNP::Dnominator |
( |
void |
| ) |
const |
|
virtual |
- Warning
- statistic undefined if there are untyped SNPs
- Returns
- Denominator of Tajima's D, or nan if there are no polymorphic sites
Definition at line 1014 of file PolySNP.cc.
◆ FuLiD()
| double Sequence::PolySNP::FuLiD |
( |
void |
| ) |
const |
|
virtual |
- Returns
- The Fu and Li (1993) D statistic, or nan if there are no polymorphic sites.
- Note
- For sequence data, an outgroup is required. This requirement is checked by assert()
- Warning
- statistic undefined if there are untyped SNPs
Definition at line 1359 of file PolySNP.cc.
◆ FuLiDStar()
| double Sequence::PolySNP::FuLiDStar |
( |
void |
| ) |
const |
|
virtual |
- Warning
- statistic undefined if there are untyped SNPs
- Returns
- Fu and Li (1993) D*, or nan if there are no polymorphic sites
Definition at line 1428 of file PolySNP.cc.
◆ FuLiF()
| double Sequence::PolySNP::FuLiF |
( |
void |
| ) |
const |
|
virtual |
- Returns
- Fu and Li (1993) F statistic, or nan if there are no polymorphic sites
- Note
- For sequence data, an outgroup is required, else undefined
- Warning
- statistic undefined if there are untyped SNPs
Definition at line 1388 of file PolySNP.cc.
◆ FuLiFStar()
| double Sequence::PolySNP::FuLiFStar |
( |
void |
| ) |
const |
|
virtual |
Fu and Li (1993) F* statistic. Incorporates correction from Simonsen et al. (1995) Genetics 141: 413, eqn A5.
- Warning
- statistic undefined if there are untyped SNPs
- Returns
- Fu and Li (1993) F* statistic, or nan if there are no polymorphic sites
Definition at line 1462 of file PolySNP.cc.
◆ Hprime()
| double Sequence::PolySNP::Hprime |
( |
const bool & |
likeThorntonAndolfatto = false | ) |
const |
|
virtual |
◆ HudsonsC()
| double Sequence::PolySNP::HudsonsC |
( |
void |
| ) |
const |
- Returns
- Hudson's (1987) estimator of
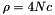 , an estimator of the population recombination rate that depends on the variance of the site frequencies. The calculation is made by a call to Recombination::HudsonsC
, an estimator of the population recombination rate that depends on the variance of the site frequencies. The calculation is made by a call to Recombination::HudsonsC
- Note
- Will return nan if there are no polymorphic sites
Definition at line 1648 of file PolySNP.cc.
◆ Minrec()
| unsigned Sequence::PolySNP::Minrec |
( |
void |
| ) |
const |
|
virtual |
- Returns
- The minimum number of recombination events observed in the sample (Hudson and Kaplan 1985). Will return SEQMAXUNSIGNED if there are < 2 segregating sites.
- Note
- Code is a modification of that provided by Jeff Wall
Definition at line 1665 of file PolySNP.cc.
◆ NumExternalMutations()
| unsigned Sequence::PolySNP::NumExternalMutations |
( |
void |
| ) |
const |
|
virtual |
- Returns
- the number of derived singletons.
- Note
- For sequence data, an outgroup is required. Will return SEQMAXUNSIGNED if that is not the case.
Definition at line 885 of file PolySNP.cc.
◆ NumMutations()
| unsigned Sequence::PolySNP::NumMutations |
( |
void |
| ) |
const |
|
virtual |
- Returns
- the total number of mutations in the data. The number of mutations per site = number of states per site - 1
Definition at line 823 of file PolySNP.cc.
◆ NumPoly()
| unsigned Sequence::PolySNP::NumPoly |
( |
void |
| ) |
const |
- Returns
- the number of polymorphic (segregating) sites in data
Definition at line 807 of file PolySNP.cc.
◆ NumSingletons()
| unsigned Sequence::PolySNP::NumSingletons |
( |
void |
| ) |
const |
|
virtual |
- Returns
- number of polymorphisms that appear once in the data, without respect to ancestral/derived
Definition at line 844 of file PolySNP.cc.
◆ SamplingVarPi()
| double Sequence::PolySNP::SamplingVarPi |
( |
void |
| ) |
const |
Component of variance of mean pairwise differences from sampling. Tajima in Takahata/Clark book, (15)
- Warning
- statistic undefined if there are untyped SNPs
Definition at line 1324 of file PolySNP.cc.
◆ StochasticVarPi()
| double Sequence::PolySNP::StochasticVarPi |
( |
void |
| ) |
const |
Stochastic variance of mean pairwise differences. Tajima in Takahata/Clark book, (14).
- Warning
- statistic undefined if there are untyped SNPs
Definition at line 1306 of file PolySNP.cc.
◆ TajimasD()
| double Sequence::PolySNP::TajimasD |
( |
void |
| ) |
const |
|
virtual |
A common summary of the site frequency spectrum. Proportional to 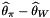 . This routine does calculate the denominator of the test statistic.
. This routine does calculate the denominator of the test statistic.
- Warning
- statistic undefined if there are untyped SNPs
- Returns
- Tajima's D, or nan if there are no polymorphic sites
- Examples:
- slidingWindow.cc, and slidingWindow2.cc.
Definition at line 929 of file PolySNP.cc.
◆ ThetaH()
| double Sequence::PolySNP::ThetaH |
( |
void |
| ) |
const |
|
virtual |
Calculate Theta ( = 4Nu) from site homozygosity, a la Fay and Wu (2000). This statistic is problematic in general to calculate when there are multiple hits. The test requires that the ancestral state (inferred from the outgroup) still be segregating in the ingroup. If that is not true, the site is skipped.
If there are >= 2 derived states inferred, a "missing data" approach is taken.
For example:
Outgroup :
A
Ingroup :
A
A
A
G
G
T
Gets treated as two sites:
A A
A A
A A
G N
G N
N T
This keeps the expectation of the statistic equal to 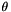 , and uses the correct number of derived mutations ovserved in the data.
, and uses the correct number of derived mutations ovserved in the data.
- Note
- When using PolySNP, an outgroup is required. When using PolySIM, which is constructed with a SimData *, an outgroup is not required (as the 0,1 coding of the data refers to ancestral and derived, respectively).
Definition at line 366 of file PolySNP.cc.
◆ ThetaL()
| double Sequence::PolySNP::ThetaL |
( |
void |
| ) |
const |
|
virtual |
Calculate Theta ( = 4Nu) from site homozygosity, corresponding to equation 1 in Thornton and Andolfatto (Genetics) "Approximate Bayesian Inference reveals evidence for a recent, severe, bottleneck in a Netherlands population of Drosophila melanogaster," (although we labelled in 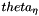 in that paper) The test requires that the ancestral state (inferred from the outgroup) still be segregating in the ingroup. If that is not true, the site is skipped.
in that paper) The test requires that the ancestral state (inferred from the outgroup) still be segregating in the ingroup. If that is not true, the site is skipped.
If there are >= 2 derived states inferred, a "missing data" approach is taken.
For example:
Outgroup :
A
Ingroup :
A
A
A
G
G
T
Gets treated as two sites:
A A
A A
A A
G N
G N
N T
This keeps the expectation of the statistic equal to , and uses the correct number of derived mutations ovserved in the data.
- Note
- For sequence data, an outgroup is required. This requirement is checked by assert()
- Author
- Joshua Shapiro
Definition at line 594 of file PolySNP.cc.
◆ ThetaPi()
| double Sequence::PolySNP::ThetaPi |
( |
void |
| ) |
const |
|
virtual |
◆ ThetaW()
| double Sequence::PolySNP::ThetaW |
( |
void |
| ) |
const |
|
virtual |
The classic "Watterson's Theta" statistic, generalized to missing data and multiple mutations per site:
![\[ \widehat\theta_w=\sum_{i=1}^{i=S}\frac{S}{\sum_{j=1}^{j=n_i-1}\frac{1}{j}} \]](form_33.png)
For this statistic, 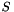 is either the number of segregating sites, or the number of mutations on the genealogy and
is either the number of segregating sites, or the number of mutations on the genealogy and 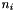 is the sample size at site i. If totMuts == 1, the number of mutations is used, else the number ofsegregating sites is used.
is the sample size at site i. If totMuts == 1, the number of mutations is used, else the number ofsegregating sites is used.
- Warning
- Statistic undefined if there are untyped SNPs. In the presence of missing data, ThetaW is calculated as the sum (over all segregating sites) of 1/a_sub_n, where a_sub_n is the denominator of ThetaW, using the number of alleles without missing data as the sample size. More formally, the routine returns a calculation base on an unweighted sample size adjustment accross sites.
Definition at line 314 of file PolySNP.cc.
◆ VarPi()
| double Sequence::PolySNP::VarPi |
( |
void |
| ) |
const |
Total variance of mean pairwise differences. Tajima in Takahata/Clark book, (13).
- Warning
- statistic undefined if there are untyped SNPs
Definition at line 1288 of file PolySNP.cc.
◆ VarThetaW()
| double Sequence::PolySNP::VarThetaW |
( |
void |
| ) |
const |
- Returns
- Variance of Watterson's Theta (ThetaW()).
- Warning
- statistic undefined if there are untyped SNPs
Definition at line 1341 of file PolySNP.cc.
◆ WallsB()
| double Sequence::PolySNP::WallsB |
( |
void |
| ) |
const |
|
virtual |
- Returns
- Wall's B Statistic. Wall, J. (1999) Genetical Research 74, pp 65-79
- Author
- Kevin Thornton
Definition at line 1144 of file PolySNP.cc.
◆ WallsBprime()
| unsigned Sequence::PolySNP::WallsBprime |
( |
void |
| ) |
const |
|
virtual |
- Returns
- Wall's B' Statistic. Wall, J. (1999) Genetical Research 74, pp 65-79
- Author
- Kevin Thornton
Definition at line 1262 of file PolySNP.cc.
◆ WallsQ()
| double Sequence::PolySNP::WallsQ |
( |
void |
| ) |
const |
|
virtual |
- Returns
- Wall's Q Statistic. Wall, J. (1999) Genetical Research 74, pp 65-79
- Author
- Kevin Thornton
Definition at line 1275 of file PolySNP.cc.
The documentation for this class was generated from the following files:
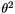 . In Thornton and Andolfatto, we simply used
. In Thornton and Andolfatto, we simply used 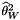 , which is slightly biased. By default, this function calculates
, which is slightly biased. By default, this function calculates 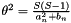 , unless this bool is set to false, in which case
, unless this bool is set to false, in which case ![\[ \widehat\theta_\pi=\sum_{i=1}^{i=S}(1-\sum_{j=1}^{j=4}\frac{k_{j,i} \times (k_{j,i}-1)}{n_i \times (n_i - 1)});k_{j,i}>0 \]](form_25.png)
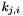 is the number of occurences of the
is the number of occurences of the 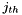 character state at site
character state at site 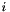 , and
, and ![\[ \widehat\theta_\pi=\sum_{i=1}^{i=S}2{p_i}{q_i} \]](form_31.png)
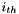 site is:
site is: