The namespace in which this library resides. More...
Namespaces | |
| Alignment | |
| Routines fundamental to aligned data. | |
| coalsim | |
| Routines for coalescent simulation. | |
| Recombination | |
| Methods dealing with recombination. | |
Classes | |
| struct | _PolySNPImpl |
| class | AlignStream |
| Virtual interface to alignment streams. More... | |
| class | AlleleCountMatrix |
| Matrix representation of allele counts in a VariantMatrix To be constructed. More... | |
| struct | AlleleCounts |
| struct | ambiguousNucleotide |
| Tests if a character is in the set A,G,C,T. More... | |
| class | ClustalW |
| ClustalW streams. More... | |
| struct | col_view_iterator |
| Iterator for column views. More... | |
| class | Comeron95 |
| Ka and Ks by Comeron's (1995) method. More... | |
| struct | ComplementBase |
| struct | countDerivedStates |
| Functor to count the number of derived states, excluding gaps and missing data, in a range of characters. More... | |
| class | Fasta |
| FASTA sequence stream. More... | |
| class | fastq |
| class | FST |
analysis of population structure using 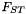 More... More... | |
| struct | GarudStats |
| class | Grantham |
| Grantham's distances. More... | |
| class | GranthamWeights2 |
| Weights paths by Grantham's distances for codons differing at 2 sites. More... | |
| class | GranthamWeights3 |
| Weights paths by Grantham's distances for codons differing at 3 sites. More... | |
| struct | HKAdata |
| Data from a single locus for an HKA test. More... | |
| struct | HKAresults |
| results of calculations of the HKA test More... | |
| struct | invalidPolyChar |
| This functor can be used to determine if a range contains characters that the SNP analysis routines in this library cannot handle gracefully. More... | |
| class | Kimura80 |
| Kimura's 2-parameter distance. More... | |
| class | nmuts |
| Calculate the number of mutations at a polymorphic site. More... | |
| struct | nSLiHS |
| struct | PairwiseLDstats |
| Pairwise linkage disequilibrium (LD) stats . More... | |
| class | phylipData |
| class | PolySIM |
| Analysis of coalescent simulation data. More... | |
| class | PolySites |
| Polymorphism tables for sequence data. More... | |
| class | PolySNP |
| Molecular population genetic analysis. More... | |
| class | PolyTable |
| The base class for polymorphism tables. More... | |
| class | PolyTableSlice |
| A container class for "sliding windows" along a polymorphism table. More... | |
| class | RedundancyCom95 |
| Calculate redundancy of a genetic code using Comeron's counting scheme. More... | |
| class | Seq |
| Abstract interface to sequence objects. More... | |
| class | shortestPath |
| Calculate shortest path between 2 codons. More... | |
| class | SimData |
| Data from coalescent simulations. More... | |
| class | SimParams |
| Parameters for Hudson's simulation program. More... | |
| class | SimpleSNP |
| SNP table data format. More... | |
| class | SingleSub |
| Deal with codons differing at 1 position. More... | |
| class | Sites |
| Calculate length statistics for divergence calculations. More... | |
| class | stateCounter |
| keep track of state counts at a site in an alignment or along a sequence More... | |
| struct | StateCounts |
| Track character state occurrence at a site in a VariantMatrix. More... | |
| class | Sums |
| class | ThreeSubs |
| Deal with codons differing at all 3 positions. More... | |
| class | TwoSubs |
| Deal with codons differing at 2 positions. More... | |
| class | Unweighted2 |
| weights all pathways equally More... | |
| class | Unweighted3 |
| weights all pathways equally More... | |
| class | VariantMatrix |
| Matrix representation of variation data. More... | |
| class | WeightingScheme2 |
| abstract interface to weighting schemes when codons differ at 2 positions More... | |
| class | WeightingScheme3 |
| abstract interface to weighting schemes when codons differ at 3 positions More... | |
Typedefs | |
| using | Com95_t = std::array< double, 19 > |
| using | Inter2_t = std::array< std::string, 2 > |
| using | Inter3_t = std::array< std::string, 9 > |
| using | polymorphicSite = std::pair< double, std::string > |
| using | polySiteVector = std::vector< polymorphicSite > |
| using | alphabet_t = std::array< const char, 16 > |
| Container type for nucleotide alphabets. | |
| typedef std::vector< std::pair< std::string, int > > | CodonUsageTable |
| using | RowView = internal::row_view_< std::int8_t * > |
| View of a VariantMatrix row (a variable site) | |
| using | ConstRowView = internal::row_view_< const std::int8_t * > |
| Const view of a VariantMatrix row (a variable site) | |
| using | ColView = internal::col_view_< std::int8_t * > |
| View of a VariantMatrix column ("haplotype") | |
| using | ConstColView = internal::col_view_< const std::int8_t * > |
| Const view of a VariantMatrix column ("haplotype") | |
Enumerations | |
| enum | GeneticCodes : std::int16_t { UNIVERSAL } |
| enum | Mutations : std::int8_t { Unknown, Ts, Tv } |
Functions | |
| template<typename T > | |
| std::istream & | operator>> (std::istream &s, AlignStream< T > &c) |
| template<typename T > | |
| std::ostream & | operator<< (std::ostream &s, const AlignStream< T > &c) |
| template<typename POINTER > | |
| col_view_iterator< POINTER > | operator+ (col_view_iterator< POINTER > i, typename col_view_iterator< POINTER >::difference_type d) |
| template<typename POINTER > | |
| col_view_iterator< POINTER > | operator+ (typename col_view_iterator< POINTER >::difference_type d, col_view_iterator< POINTER > i) |
| template<typename POINTER > | |
| col_view_iterator< POINTER > & | operator++ (col_view_iterator< POINTER > &i) |
| template<typename POINTER > | |
| col_view_iterator< POINTER > | operator- (col_view_iterator< POINTER > i, typename col_view_iterator< POINTER >::difference_type d) |
| template<typename POINTER > | |
| col_view_iterator< POINTER > | operator- (typename col_view_iterator< POINTER >::difference_type d, col_view_iterator< POINTER > i) |
| template<typename POINTER > | |
| col_view_iterator< POINTER > & | operator-- (col_view_iterator< POINTER > &i) |
| template<typename POINTER > | |
| col_view_iterator< POINTER > & | operator+= (col_view_iterator< POINTER > &i, typename col_view_iterator< POINTER >::difference_type d) |
| template<typename POINTER > | |
| col_view_iterator< POINTER > & | operator-= (col_view_iterator< POINTER > &i, typename col_view_iterator< POINTER >::difference_type d) |
| template<typename POINTER > | |
| col_view_iterator< POINTER >::difference_type | operator- (col_view_iterator< POINTER > i, col_view_iterator< POINTER > j) |
| CodonUsageTable | makeCodonUsageTable (const Seq *sequence) |
| CodonUsageTable | makeCodonUsageTable (const std::string &sequence) |
| CodonUsageTable | makeCodonUsageTable (std::string::const_iterator beg, std::string::const_iterator end) |
| Mutations | TsTv (const char &i, const char &j) |
| Mutations | TsTv (const int &i, const int &j) |
| bool | Different (const std::string &seq1, const std::string &seq2, const bool &skip_missing=true, const bool &nucleic_acid=true) |
| int | NumDiffs (const std::string &seq1, const std::string &seq2, const bool &skip_missing=true, const bool &nucleic_acid=true) |
| bool | Gapped (const std::string &s) |
| template<typename Iterator > | |
| bool | Gapped (Iterator beg, Iterator end, const char &gapchar='-') |
| bool | NotAGap (const char &c) |
| template<typename key , typename value > | |
| std::vector< std::pair< key, value > > | operator+ (const std::vector< std::pair< key, value > > &lhs, const std::vector< std::pair< key, value > > &rhs) |
| template<typename key , typename value > | |
| std::vector< std::pair< key, value > > | operator+= (std::vector< std::pair< key, value > > &lhs, const std::vector< std::pair< key, value > > &rhs) |
| template<typename key , typename value , typename comparison > | |
| std::map< key, value, comparison > | operator+ (const std::map< key, value, comparison > &lhs, const std::map< key, value, comparison > &rhs) |
| template<typename key , typename value , typename comparison > | |
| std::map< key, value, comparison > | operator+= (std::map< key, value, comparison > &lhs, const std::map< key, value, comparison > &rhs) |
| template<typename iterator > | |
| double | mean (iterator beg, iterator end) |
| template<typename iterator > | |
| double | variance (iterator beg, iterator end) |
| template<typename ForwardIterator > | |
| std::pair< double, double > | meanAndVar (ForwardIterator beg, ForwardIterator end) |
| template<typename T > | |
| const Sums< T > | operator+ (const Sums< T > &lhs, const Sums< T > &rhs) |
| template<typename T > | |
| const Sums< T > | operator+ (const Sums< T > &lhs, const T &rhs) |
| class | __attribute__ ((deprecated)) countStates |
| Functor to count the number of states, excluding gaps and missing data, in a range of characters. More... | |
| HKAresults | calcHKA (const std::vector< HKAdata > &data) |
| Inter2_t | Intermediates2 (const std::string &codon1, const std::string &codon2) |
| Calculate the intermediate codons between a pair of codons diverged at 2 positions. More... | |
| Inter3_t | Intermediates3 (const std::string &codon1, const std::string &codon2) |
| Calculate the intermediate codons between a pair of codons diverged at 3 positions. More... | |
| polySiteVector | make_polySiteVector (const Sequence::PolyTable &data) __attribute__((deprecated)) |
| std::istream & | operator>> (std::istream &s, PolyTable &c) |
| std::ostream & | operator<< (std::ostream &o, const PolyTable &c) |
| bool | containsCharacter (const PolyTable *t, const char ch) __attribute__((deprecated)) |
| bool | polyTableValid (const PolyTable *t) __attribute__((deprecated)) |
| template<typename T > | |
| T | copyPolyTable (const T &t) |
| template<typename T , typename F > | |
| T | removeColumns (const T &t, const F &f, const bool skipAnc=false, const unsigned anc=0, const char gapchar='-') __attribute__((deprecated)) |
| template<typename T > | |
| T | removeGaps (const T &t, const bool skipAnc=false, const unsigned anc=0, const char gapchar='-') __attribute__((deprecated)) |
| template<typename T > | |
| T | removeInvariantPos (const T &t, const bool skipAnc=false, const unsigned anc=0, const char gapchar='-') __attribute__((deprecated)) |
| template<typename T > | |
| T | removeAmbiguous (const T &t, const bool skipAnc=false, const unsigned anc=0, const char gapchar='-') __attribute__((deprecated)) |
| template<typename T > | |
| T | removeMissing (const T &t, const bool skipAnc=false, const unsigned anc=0, const char gapchar='-') __attribute__((deprecated)) |
| template<typename T > | |
| T | removeMultiHits (const T &t, const bool skipAnc=false, const unsigned anc=0, const char gapchar='-') __attribute__((deprecated)) |
| template<typename T > | |
| T | polyTableToBinary (const T &t, const unsigned ref=0, const char gapchar='-') __attribute__((deprecated)) |
| template<typename T > | |
| T | polyTableFreqFilter (const T &t, const unsigned mincount, const bool skipAnc=false, const unsigned anc=0, const char gapchar='-') __attribute__((deprecated)) |
| std::ostream & | operator<< (std::ostream &s, const Seq &c) |
| std::istream & | operator>> (std::istream &s, Seq &c) |
| bool | isDNA (const char &ch) |
| test if character is part of Sequence::dna_alphabet More... | |
| template<typename Iter > | |
| bool | validSeq (Iter beg, Iter end, const char *_pattern=Sequence::basic_dna_alphabet, const bool icase=true) |
| template<typename Iterator > | |
| std::map< typename std::iterator_traits< Iterator >::value_type, unsigned > | makeCountList (Iterator beg, Iterator end) |
| template<typename Iterator > | |
| bool | internalGapCheck (Iterator beg, Iterator end, const char &gapchar='-', const unsigned &mod=3) |
| std::pair< unsigned, unsigned > | mutsShortestPath (const std::string &codon1, const std::string &codon2, const Sequence::GeneticCodes &code=GeneticCodes::UNIVERSAL) |
| std::pair< unsigned, shortestPath::pathType > | diffType (const std::string &codon1, const std::string &codon2, const Sequence::GeneticCodes &code=GeneticCodes::UNIVERSAL) |
| std::tuple< shortestPath::pathType, shortestPath::pathType, shortestPath::pathType > | diffTypeMulti (const std::string &codon1, const std::string &codon2, const Sequence::GeneticCodes &code=GeneticCodes::UNIVERSAL) |
| std::istream & | operator>> (std::istream &s, SimParams &c) |
| std::vector< StateCounts > | process_variable_sites (const VariantMatrix &m, const std::vector< std::int8_t > &refstates) |
| std::vector< StateCounts > | process_variable_sites (const VariantMatrix &m, const std::int8_t refstate) |
| std::vector< StateCounts > | process_variable_sites (const VariantMatrix &m) |
| GarudStats | H1H12 (const SimData &d) __attribute__((deprecated)) |
| std::vector< double > | lHaf (const SimData &data, const double l) |
| std::pair< double, double > | nSL (const std::size_t &core, const SimData &d, const std::unordered_map< double, double > &gmap=std::unordered_map< double, double >()) __attribute__((deprecated)) |
| double | Snn_statistic (const unsigned individuals[], const std::vector< std::vector< double > > &dkj, const unsigned config[], const size_t &npop, const unsigned &nsam) __attribute__((deprecated)) |
| template<typename shuffler > | |
| std::pair< double, double > | Snn_test (const PolyTable &snpTable, const unsigned config[], const size_t &npop, shuffler &s, const unsigned &nperms=10000) __attribute__((deprecated)) |
| template<typename shuffler > | |
| std::vector< std::vector< double > > | Snn_test_pairwise (const PolyTable &snpTable, const unsigned config[], const size_t &npop, shuffler &s, const unsigned &nperms=10000) __attribute__((deprecated)) |
| std::vector< AlleleCounts > | allele_counts (const AlleleCountMatrix &m) |
| Count number of alleles at each site. More... | |
| std::vector< AlleleCounts > | non_reference_allele_counts (const AlleleCountMatrix &m, const std::int8_t refstate) |
| Count number of non-reference alleles at each site. More... | |
| std::vector< AlleleCounts > | non_reference_allele_counts (const AlleleCountMatrix &m, const std::vector< std::int8_t > &refstates) |
| Count number of non-reference alleles at each site. More... | |
| double | tajd (const AlleleCountMatrix &ac) |
| Tajima's D. More... | |
| double | hprime (const AlleleCountMatrix &ac, const std::int8_t refstate) |
| double | hprime (const AlleleCountMatrix &m, const std::vector< std::int8_t > &refstates) |
| double | faywuh (const AlleleCountMatrix &ac, const std::int8_t refstate) |
| Fay and Wu's H. More... | |
| double | faywuh (const AlleleCountMatrix &ac, const std::vector< std::int8_t > &refstates) |
| Fay and Wu's H. More... | |
| std::vector< std::int32_t > | difference_matrix (const VariantMatrix &m) |
| Calculate number of differences between all samples. More... | |
| std::vector< std::int32_t > | is_different_matrix (const VariantMatrix &m) |
| std::vector< std::int32_t > | label_haplotypes (const VariantMatrix &m) |
| Assign a unique label to each haplotype. More... | |
| std::int32_t | number_of_haplotypes (const VariantMatrix &m) |
| Calculate the number of haplotypes in a sample. More... | |
| double | haplotype_diversity (const VariantMatrix &m) |
| Calculate the haplotype diversity of a sample. More... | |
| std::int32_t | rmin (const VariantMatrix &m) |
| GarudStats | garud_statistics (const VariantMatrix &m) |
| Calculate H1, H12, and H2/H1. More... | |
| double | diversity_from_counts (const std::unordered_map< std::int32_t, std::int32_t > &counts, const std::size_t nsam) |
| Calculate heterozygosity/diversity from count data. More... | |
| std::vector< TwoLocusCounts > | two_locus_haplotype_counts (const VariantMatrix &m, std::size_t sitei, const std::size_t sitej, const bool skip_missing) |
| nSLiHS | nsl (const VariantMatrix &m, const std::size_t core, const std::int8_t refstate) |
| nSL and iHS statistics More... | |
| std::uint32_t | nvariable_sites (const AlleleCountMatrix &m) |
| Number of polymorphic sites. More... | |
| std::uint32_t | nbiallelic_sites (const AlleleCountMatrix &m) |
| Number of bi-allelic sites. More... | |
| std::uint32_t | total_number_of_mutations (const AlleleCountMatrix &m) |
| Total number of mutations in the sample. More... | |
| double | thetah (const AlleleCountMatrix &ac, const std::int8_t refstate) |
Fay and Wu's 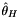 . More... . More... | |
| double | thetah (const AlleleCountMatrix &m, const std::vector< std::int8_t > &refstates) |
| Fay and Wu's . More... | |
| double | thetal (const AlleleCountMatrix &ac, const std::int8_t refstate) |
Zeng et al. 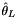 . More... . More... | |
| double | thetal (const AlleleCountMatrix &m, const std::vector< std::int8_t > &refstates) |
| Zeng et al. . More... | |
| double | thetapi (const AlleleCountMatrix &ac) |
| Mean pairwise differences. More... | |
| double | thetaw (const AlleleCountMatrix &ac) |
| Watterson's theta. More... | |
| template<typename T > | |
| bool | all_missing (const T &t) |
| std::string | Translate (std::string::const_iterator beg, std::string::const_iterator end, Sequence::GeneticCodes genetic_code=GeneticCodes::UNIVERSAL, const char &gapchar='-') |
| std::int32_t | filter_sites (VariantMatrix &m, const std::function< bool(const RowView &)> &f) |
| std::int32_t | filter_haplotypes (VariantMatrix &m, const std::function< bool(const ColView &)> &f) |
| template<typename streamtype > | |
| VariantMatrix | from_msformat (streamtype &input_stream) |
| Create VariantMatrix from "ms"-like input format. More... | |
| template<typename output_stream > | |
| void | to_msformat (const VariantMatrix &m, output_stream &o) |
| Write VariantMatrix in "ms" format. More... | |
| VariantMatrix | make_window (const VariantMatrix &m, const double beg, const double end) |
| Return a window from a VariantMatrix. More... | |
| VariantMatrix | make_slice (const VariantMatrix &m, const double beg, const double end, const std::size_t i, const std::size_t j) |
| Return a slice from a VariantMatrix. More... | |
| ConstRowView | get_RowView (const VariantMatrix &m, const std::size_t row) |
Return a ConstRowView from VariantMatrix m at row row. std::out_of_range is thrown if row is out of range. | |
| RowView | get_RowView (VariantMatrix &m, const std::size_t row) |
| ConstRowView | get_ConstRowView (const VariantMatrix &m, const std::size_t row) |
Return a ConstRowView from VariantMatrix m at row row. std::out_of_range is thrown if row is out of range. | |
| ConstRowView | get_ConstRowView (VariantMatrix &m, const std::size_t row) |
Return a ConstRowView from VariantMatrix m at row row. std::out_of_range is thrown if row is out of range. | |
| ColView | get_ColView (VariantMatrix &m, const std::size_t col) |
Return a ColView from VariantMatrix m at col col. std::out_of_range is thcoln if col is out of range. | |
| ConstColView | get_ColView (const VariantMatrix &m, const std::size_t col) |
Return a ConstColView from VariantMatrix m at col col. std::out_of_range is thcoln if col is out of range. | |
| ConstColView | get_ConstColView (VariantMatrix &m, const std::size_t col) |
Return a ConstColView from VariantMatrix m at col col. std::out_of_range is thcoln if col is out of range. | |
| ConstColView | get_ConstColView (const VariantMatrix &m, const std::size_t col) |
Return a ConstColView from VariantMatrix m at col col. std::out_of_range is thcoln if col is out of range. | |
| polySiteVector | rotatePolyTable (const Sequence::PolyTable *data) |
| std::ostream & | operator<< (std::ostream &stream, class SimParams &object) |
| set< string > | uhaps (d.begin(), d.end()) |
| vector< string > | vuhaps (uhaps.size()) |
| hapcounts | reserve (uhaps.size()) |
| for (auto &uh :uhaps) | |
| for (auto c :hapcounts) | |
| sort (hapcounts.begin(), hapcounts.end(), std::bind(greater< double >(), std::placeholders::_1, std::placeholders::_2)) | |
| return | GarudStats (H1, H12, H2H1) |
| double | Dij (const polymorphicSite &p, const std::vector< unsigned > &config, const unsigned &i, const unsigned &j) |
| double | Gmin (const polySiteVector &, const std::vector< unsigned > &) |
| void | fst (const VariantMatrix &m, const std::vector< std::pair< std::size_t, std::size_t >> &partitions) |
| nSLiHS | nslx (const VariantMatrix &m, const std::size_t core, const std::int8_t refstate, const int x) |
| PairwiseLDStats | pairwise_ld_details (const VariantMatrix &m, const std::size_t i, const std::size_t j, const std::int8_t refstate) |
| std::vector< PairwiseLDStats > | pairwise_ld (const Sequence::VariantMatrix &m, const std::int8_t refstate) |
| template<typename T > | |
| std::int32_t | filter_common (VariantMatrix &m, const std::function< bool(const T &)> &f, const std::function< T(VariantMatrix &, const std::size_t)> &viewmaker, std::size_t &dim) |
Variables | |
| class __attribute__((deprecated)) SimpleSNP typedef SimpleSNP | Hudson2001 |
| const alphabet_t | dna_alphabet |
| Alphabet for DNA sequences Valid DNA characters. Upper-case only. Only - is accepted as gap characters. More... | |
| const alphabet_t | dna_poly_alphabet |
| Alphabet for polymorphism (SNP) analysis. 16 characters are used so that we may encode 2 nucleotides in a 8-bit integer. More... | |
| const alphabet_t::size_type | NOTPOLYCHAR = dna_poly_alphabet.size() |
| An index from dna_poly_alphabet >= this is not a valid character for variation analysis. | |
| const alphabet_t::size_type | POLYEOS |
| The value of terminating an encoded string of SNP data. More... | |
| const unsigned | SEQMAXUNSIGNED = std::numeric_limits<unsigned>::max() |
| const double | SEQMAXDOUBLE = std::numeric_limits<double>::max() |
| const char * | basic_dna_alphabet = "[^AGTCN\\-]" |
| const char * | full_dna_alphabet = "[^AGCTNXMRWSKVHDB\\-]" |
| const char * | pep_alphabet = "[^ARNDBCQEZGHILKMFPSTWYV\\-]" |
| GarudStats | |
| vector< double > | hapcounts |
| const double | denom = static_cast<double>(d.size() * (d.size() - 1)) |
| double | H1 = 0.0 |
| double | H12 |
| double | H2H1 |
Detailed Description
The namespace in which this library resides.
The entirety of this library is defined in namespace Sequence.
Typedef Documentation
◆ CodonUsageTable
| typedef std::vector< std::pair<std::string,int> > Sequence::CodonUsageTable |
A CodonUsageTable is a vector of pairs. In each pair, the first element is the codon, and the second element is an integer counting the number of occurrences of the codon
Definition at line 43 of file typedefs.hpp.
◆ polymorphicSite
| using Sequence::polymorphicSite = typedef std::pair< double, std::string > |
For polymorphism data, a Site can be represented as a position (a double) and the characters at that positions (a std::string)
Definition at line 45 of file polySiteVector.hpp.
◆ polySiteVector
| using Sequence::polySiteVector = typedef std::vector< polymorphicSite > |
A polymorphism data set can be represented as a vector containing a sequence of polymorphicSite
Definition at line 51 of file polySiteVector.hpp.
Enumeration Type Documentation
◆ GeneticCodes
|
strong |
Only UNIVERSAL (= 0) is currently supported. The order of the genetic codes is that of NCBI's code tables, available at http://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi
Definition at line 40 of file SeqEnums.hpp.
◆ Mutations
|
strong |
Values: Unknown=0,Ts, and Tv.
Unknown means unknown, Ts means transition, Tv means transversion
Definition at line 45 of file SeqEnums.hpp.
Function Documentation
◆ __attribute__()
| class Sequence::__attribute__ | ( | (deprecated) | ) |
Functor to count the number of states, excluding gaps and missing data, in a range of characters.
- Parameters
-
beg beginning of a range of characters end end of a range of characters haveOutgroup true of one of the elements of site is an outgroup state, false otherwise outgroup the index of the outgroup sequence in site
- Note
- will return an empty object if beg>=end or beg+outgroup>=end
- Parameters
-
_ssh a value of ssh to increment site an object representing the value type of PolyTable::const_site_iterator haveOutgroup true of one of the elements of site is an outgroup state, false otherwise outgroup the index of the outgroup sequence in site
Data type to store site positions
Data type for storing genotypes
non-const reference to std::string
const reference to std::string
The size_type for the haplotype vector
non-const iterator to the haplotypes
const iterator to the haplotypes
non-const iterator to the positions
const iterator to the positions
Const iterator to segregating sites Const iterator to segregating sites. The value_type of this iterator is const std::pair<double,std::string>, where the double is the position of the segregating site, and the string the list of states at the site. The first character in the string corresponds to the state of the first character in the PolyTable (i.e. (*this)[0]), etc.
- Returns
- an iterator pointing to the first "haplotype"
- an iterator pointing the end of the "haplotypes"
- a const iterator pointing to the first "haplotype"
- a const iterator pointing the end of the "haplotypes"
- a const iterator pointing to the first "haplotype"
- a const iterator pointing the end of the "haplotypes"
- iterator to first position
- iterator to end of positions
- const iterator to first position
- const iterator to end of positions
- const iterator to first position
- const iterator to end of positions
- const iterator to first column (position, variants)
- const iterator to end of columns
- const iterator to first column (position, variants)
- const iterator to first column (position, variants)
Convenience function to return site positions
Conventience function to return data. Each string is a "haplotype".
Comparison operator. Case-sensitive
Not-equal operator. Case-sensitive
Move assignment
Copy assignment
Return the i-th element of PolyTable::data.
- Note
- range-checking done by assert()
Return the i-th element of PolyTable::data.
- Note
- range-checking done by assert()
- Returns
- true if object contains no data, false otherwise
Assignment operation, allowing a range of polymorphic sites to be assigned to a polymorphism table. This exists mainly for two purposes. One is the ability to assign tables from "slices" of other tables. Second is to facilitate the writing of "sliding window" routines.
- Returns
- true if the assignment was successful, false otherwise. The only case where false is returned is if the number of individuals at each site is not the constant from beg to end.
Assign data to object
- Returns
- true if successful
Move data to object
- Returns
- true if successful
- Sample size
- the i-th position from the PolyTable::positions.
- the number of positions (columns)
read is a pure virtual function. Calls to istream & operator>> (istream & s, PolyTable & c) act via this routine, which must be defined in all derived classes
print is a pure virtual function. Calls to ostream & operator<<(ostream & s, PolyTable & c) act via this routine, which must be defined in all derived classes
- Parameters
-
beg beginning of a range of characters end end of a range of characters haveOutgroup true of one of the elements of site is an outgroup state, false otherwise outgroup the index of the outgroup sequence in site
- Note
- will return an empty object if beg>=end or beg+outgroup>=end
- Parameters
-
_ssh a value of ssh to increment site an object representing the value type of PolyTable::const_site_iterator haveOutgroup true of one of the elements of site is an outgroup state, false otherwise outgroup the index of the outgroup sequence in site
Data type to store site positions
Data type for storing genotypes
non-const reference to std::string
const reference to std::string
The size_type for the haplotype vector
non-const iterator to the haplotypes
const iterator to the haplotypes
non-const iterator to the positions
const iterator to the positions
Const iterator to segregating sites Const iterator to segregating sites. The value_type of this iterator is const std::pair<double,std::string>, where the double is the position of the segregating site, and the string the list of states at the site. The first character in the string corresponds to the state of the first character in the PolyTable (i.e. (*this)[0]), etc.
- Returns
- an iterator pointing to the first "haplotype"
- an iterator pointing the end of the "haplotypes"
- a const iterator pointing to the first "haplotype"
- a const iterator pointing the end of the "haplotypes"
- a const iterator pointing to the first "haplotype"
- a const iterator pointing the end of the "haplotypes"
- iterator to first position
- iterator to end of positions
- const iterator to first position
- const iterator to end of positions
- const iterator to first position
- const iterator to end of positions
- const iterator to first column (position, variants)
- const iterator to end of columns
- const iterator to first column (position, variants)
- const iterator to first column (position, variants)
Convenience function to return site positions
Conventience function to return data. Each string is a "haplotype".
Comparison operator. Case-sensitive
Not-equal operator. Case-sensitive
Move assignment
Copy assignment
Return the i-th element of PolyTable::data.
- Note
- range-checking done by assert()
Return the i-th element of PolyTable::data.
- Note
- range-checking done by assert()
- Returns
- true if object contains no data, false otherwise
Assignment operation, allowing a range of polymorphic sites to be assigned to a polymorphism table. This exists mainly for two purposes. One is the ability to assign tables from "slices" of other tables. Second is to facilitate the writing of "sliding window" routines.
- Returns
- true if the assignment was successful, false otherwise. The only case where false is returned is if the number of individuals at each site is not the constant from beg to end.
Assign data to object
- Returns
- true if successful
Move data to object
- Returns
- true if successful
- Sample size
- the i-th position from the PolyTable::positions.
- the number of positions (columns)
read is a pure virtual function. Calls to istream & operator>> (istream & s, PolyTable & c) act via this routine, which must be defined in all derived classes
print is a pure virtual function. Calls to ostream & operator<<(ostream & s, PolyTable & c) act via this routine, which must be defined in all derived classes
◆ all_missing()
|
inline |
Returns true if all elements in t encode missing data. T should be a model of a VariantMatrix, RowView, or ColumnView
◆ containsCharacter()
| bool Sequence::containsCharacter | ( | const PolyTable * | t, |
| const char | ch | ||
| ) |
- Returns
- true if t contains ch, false otherwise
Definition at line 33 of file PolyTableFunctions.cc.
◆ Different()
| bool Sequence::Different | ( | const std::string & | seq1, |
| const std::string & | seq2, | ||
| const bool & | skip_missing, | ||
| const bool & | nucleic_acid | ||
| ) |
Ask if two strings are different. While this can normally be done by asking if (seq1 != seq2) {}, missing data poses a problem here. If skip-missing == 1, missing data (the 'N' character for nucleotide data, 'X' for amino acid) are not used to determine if the sequences are different. If nucleic_acid ==1, nucleotide data are assumed, if nucleic_acid==0, protein data are assumed.
- Note
- case-insensitive
- Returns
- true if the seqs are different, false otherwise. If the two sequences are of different length, true is returned.
Definition at line 98 of file Comparisons.cc.
◆ diffType()
| std::pair< unsigned, shortestPath::pathType > Sequence::diffType | ( | const std::string & | codon1, |
| const std::string & | codon2, | ||
| const Sequence::GeneticCodes & | code | ||
| ) |
- Parameters
-
codon1 a std::string of length 3 representing a codon codon2 a std::string of length 3 representing a codon code the genetic code to use in translating the codons
- Returns
- a std::pair<unsigned,shortestPath::pathType>. The first member of the pair takes a value of either 0,1, or 2, depending on the site at which the two codons differ (1st, 2nd, or 3rd position, respectively). If the codons differ at more than 1 site, or contain characters other that {A,G,C,T}, the first member will be set to Sequence::SEQMAXUNSIGNED. The second member will have the value Sequence::shortestPath::pathType::N if the change is nonsynonymous, Sequence::shortestPath::pathType::S if synonymous, Sequence::shortestPath::shortestPath::pathType::NONE if the codons don't differ, and Sequence::shortestPath::pathType::AMBIG if any of the codons contain characters other than {A,G,C,T}.
- Precondition
- (codon1.length()==3 && codon2.length() == 3)
Definition at line 425 of file shortestPath.cc.
◆ diffTypeMulti()
| std::tuple< shortestPath::pathType, shortestPath::pathType, shortestPath::pathType > Sequence::diffTypeMulti | ( | const std::string & | codon1, |
| const std::string & | codon2, | ||
| const Sequence::GeneticCodes & | code | ||
| ) |
- Returns
- a tuple representing the type of single position changes between codon1 and codon2. There is one value in the tuple for each codon position.
- Note
- The values are assigned as follows: For each position in codon1, and 2, swap the i-th state between the two codons. If this results in a replacement change in both cases, record shortestPath::N. If it's synonymous in both cases, record shortestPath::S. If the swap results in no change at all (i.e. the two bases are identical), record shortestPath::pathType::NONE. For all other cases, record shortestPath::AMBIG. This function is most useful at identifying mutations that can be unambiguously classifies as silent or replacement. Note that, if one considers the pathways possible between codons, all sites can be assigned as N or S. For such applications, use Sequence::shortestPath.
Definition at line 478 of file shortestPath.cc.
◆ diversity_from_counts()
| double Sequence::diversity_from_counts | ( | const std::unordered_map< std::int32_t, std::int32_t > & | counts, |
| const std::size_t | nsam | ||
| ) |
Calculate heterozygosity/diversity from count data.
- Parameters
-
counts a vector counts. nsam the sample size
- Returns
- diversity = 1 - homozygosity
Definition at line 10 of file generic.cc.
◆ Gapped() [1/2]
| bool Sequence::Gapped | ( | const std::string & | s | ) |
Ask if the std::string contains a gap character.
- Returns
- true if the string contains gaps, false otherwise
- Note
- The only gap character checked so far is '-'. Use template version for other gap characters
Definition at line 184 of file Comparisons.cc.
◆ Gapped() [2/2]
| bool Sequence::Gapped | ( | Iterator | beg, |
| Iterator | end, | ||
| const char & | gapchar = '-' |
||
| ) |
- Parameters
-
beg an iterator end an iterator gapchar a character representing an aligment gap
- Returns
- true if gapchar is present in the range [beg,end), false otherwise
Definition at line 58 of file Comparisons.hpp.
◆ garud_statistics()
| GarudStats Sequence::garud_statistics | ( | const VariantMatrix & | m | ) |
◆ internalGapCheck()
| bool Sequence::internalGapCheck | ( | Iterator | beg, |
| Iterator | end, | ||
| const char & | gapchar = '-', |
||
| const unsigned & | mod = 3 |
||
| ) |
This function checks a range for internal gaps that meet a certain length requirement. The requirement is that lengthmod == 0. The value true is returned if this is not the case, false otherwise. One use of this function may be to check that the internal gaps in an aligned cds sequence are all multiples of 3 in length.
Definition at line 86 of file SeqUtilities.hpp.
◆ is_different_matrix()
| std::vector< std::int32_t > Sequence::is_different_matrix | ( | const VariantMatrix & | m | ) |
Returns whether or not haplotypes differ.
- Parameters
-
m A VariantMatrix
- Returns
- std::vector<std::int32_t>
This function is conceptually indentical to difference_matrix, but the return value contains 0 = identical, 1 = different rather than the actual number of differences. Thus, it is faster to calculate when the binary answer is needed.
Definition at line 45 of file haplotype_statistics.cc.
◆ make_slice()
| VariantMatrix Sequence::make_slice | ( | const VariantMatrix & | m, |
| const double | beg, | ||
| const double | end, | ||
| const std::size_t | i, | ||
| const std::size_t | j | ||
| ) |
Return a slice from a VariantMatrix.
- Parameters
-
m A VariantMatrix beg Beginning of window end End of window i index of first haplotype to include j one past last haplotype to include
The result is a variant matrix including positions [beg,end] and samples [i,j) from m. Note that the sample interval is half-open!
Definition at line 34 of file windows.cc.
◆ make_window()
| VariantMatrix Sequence::make_window | ( | const VariantMatrix & | m, |
| const double | beg, | ||
| const double | end | ||
| ) |
Return a window from a VariantMatrix.
- Parameters
-
m A VariantMatrix beg Beginning of window end End of window
- Note
- The window intervals are open, [beg,end]
Definition at line 6 of file windows.cc.
◆ makeCodonUsageTable() [1/3]
| CodonUsageTable Sequence::makeCodonUsageTable | ( | const Seq * | sequence | ) |
- Parameters
-
sequence and object of type Sequence::Seq1
- Returns
- and object of type Sequence::CodonUsageTable
- Note
- Assumes first character of sequence is a first codon position.
- Examples:
- codons.cc.
Definition at line 81 of file CodonTable.cc.
◆ makeCodonUsageTable() [2/3]
| CodonUsageTable Sequence::makeCodonUsageTable | ( | const std::string & | sequence | ) |
- Parameters
-
sequence and object of type std::string
- Returns
- and object of type Sequence::CodonUsageTable
- Note
- Assumes first character of sequence is a first codon position
Definition at line 91 of file CodonTable.cc.
◆ makeCodonUsageTable() [3/3]
| CodonUsageTable Sequence::makeCodonUsageTable | ( | std::string::const_iterator | beg, |
| std::string::const_iterator | end | ||
| ) |
- Parameters
-
beg a const_iterator to the beginning of a std::string or Sequence::Seq end a const_iterator to the end of a std::string or Sequence::Seq
- Returns
- and object of type Sequence::CodonUsageTable
- Note
- beg and end can be adjusted to point to the first at last positions in a CDS
Definition at line 101 of file CodonTable.cc.
◆ makeCountList()
| std::map< typename std::iterator_traits< Iterator >::value_type, unsigned > Sequence::makeCountList | ( | Iterator | beg, |
| Iterator | end | ||
| ) |
- Parameters
-
beg an iterator end an iterator
- Returns
- a std::map< type, unsigned >, where type is the iterator_traits<Iterator>::value_type of Iterator. The keys are the (unique) elements present in the range, and the unsinged values the numbers of times each element occurs occur
- Note
- This function can be used as an alternative to Sequence::stateCounter if you want to count more than just strict DNA characters.
Definition at line 61 of file SeqUtilities.hpp.
◆ meanAndVar()
| std::pair<double,double> Sequence::meanAndVar | ( | ForwardIterator | beg, |
| ForwardIterator | end | ||
| ) |
A function to calculate the mean and variance of the values stored in a container. The rationale is that when both the mean and the variance (an sum of squares) are needed, it is more efficient to calculate them together, because you only go over the data once.
◆ NotAGap()
| bool Sequence::NotAGap | ( | const char & | c | ) |
- Returns
- true if a c is not a gap character, false otherwise.
- Note
- Currently, only '-' is considered to be a gap character
Definition at line 196 of file Comparisons.cc.
◆ NumDiffs()
| int Sequence::NumDiffs | ( | const std::string & | seq1, |
| const std::string & | seq2, | ||
| const bool & | skip_missing, | ||
| const bool & | nucleic_acid | ||
| ) |
- Parameters
-
seq1 A string representing a sequence seq2 A string representing a sequence skip_missing If true, missing data characters will not be counted as differences nucleic_acid. If true, n/N are the missing data symbol. If false, x/X.
- Returns
- the number of differences between two std::strings. Can skip missing data in the same fashion as Comparisons::Different. If one sequence is shorter than the other, -1 is returned
Definition at line 141 of file Comparisons.cc.
◆ polyTableValid()
| bool Sequence::polyTableValid | ( | const PolyTable * | t | ) |
- Returns
- true if the following conditions are met : First, the length of every row in the table is equal to the length of the vector of positions. Second, all of the characters in the data are members of the set {A,G,C,T,N,-} (case-insensitive).
This function is useful if you play around with PolyTable objects in non-const contexts, or read them in from files and need to check that the data are compatible with other routines in this library. This routine can be thought of as a PolyTable equivalent to Alignment::validForPolyAnalysis, which works on ranges of Sequence::Seq objects.
Definition at line 48 of file PolyTableFunctions.cc.
◆ Snn_statistic()
| double Sequence::Snn_statistic | ( | const unsigned | individuals[], |
| const std::vector< std::vector< double > > & | dkj, | ||
| const unsigned | config[], | ||
| const size_t & | npop, | ||
| const unsigned & | nsam | ||
| ) |
◆ TsTv() [1/2]
| Mutations Sequence::TsTv | ( | const char & | i, |
| const char & | j | ||
| ) |
Takes two chars, assumed to be nucleotides. The integer returned by this function is a member of the enumeration type Sequence::Mutations.
Definition at line 35 of file Comparisons.cc.
◆ TsTv() [2/2]
| Mutations Sequence::TsTv | ( | const int & | i, |
| const int & | j | ||
| ) |
Takes two ints, assumed to be integer representations of nucleotides. The conversion of int to nucleotide is via Sequence::dna_alphabet
Definition at line 71 of file Comparisons.cc.
◆ validSeq()
| bool Sequence::validSeq | ( | Iter | beg, |
| Iter | end, | ||
| const char * | _pattern = Sequence::basic_dna_alphabet, |
||
| const bool | icase = true |
||
| ) |
- Parameters
-
beg an iterator to the beginning of a range end an iterator to the end of a range _pattern the (complement of the) alphabet as a regular expression. icase defaults to case insensitive matching. Pass "false" to make matching case sensitive The character set is complemented because we test for not in the alphabet
- Returns
- true if beg and end define a range of valid characters. The range is valid if and only if all characters in the range are present in the pattern (i.e. are not part of the set of characters that complement the pattern)
- Examples:
- valid_dna.cc.
Definition at line 55 of file SeqRegexes.hpp.
Variable Documentation
◆ basic_dna_alphabet
| const char* Sequence::basic_dna_alphabet = "[^AGTCN\\-]" |
A regex for the complement of the minimal DNA alphabet
Definition at line 40 of file SeqRegexes.hpp.
◆ full_dna_alphabet
| const char* Sequence::full_dna_alphabet = "[^AGCTNXMRWSKVHDB\\-]" |
A regex for the complement of a complete DNA alphabet
- Examples:
- valid_dna.cc.
Definition at line 45 of file SeqRegexes.hpp.
◆ GarudStats
H1 is total haplotype homozygosity. H2 is haplotype homozygosity, combining two most common haplotypes. H2 = H1 + 2p1p2 H2H1 = H2/H1, where H2 is haplotype homozygosity for all but most common haplotype. H2H1 = (H1 - p1^2)/H1
◆ H12
| double Sequence::H12 |
◆ H2H1
| double Sequence::H2H1 |
◆ Hudson2001
| class __attribute__ ((deprecated)) SimpleSNP typedef SimpleSNP Sequence::Hudson2001 |
The two bools that this constructor takes allow you to deal with two very different types of polymorphism data. If both bools are set to 0 (the default), the data are simply read in as they are. However, if diploid == 1, then if n sequences are read in, the data are assumed to be diploid (make sense...), and are converted into 2n strings. Further, if heterozygous bases are encoutered (R,W, etc.), the two possible states are arbitrarily assigned to each sequence.
If isofemale==1, it is assumed that the data represent real haplotype data (i.e. phase is known). The name for the bool comes from the fact that data gathered from Drosophila lines is often obtained from isofemale stocks, making them homozygous such that the phase of each SNP is known. If a heterozygous base is found, one of the two possible states will be assigned randomly (NOT IMPLEMENTED YET!)
Definition at line 67 of file SimpleSNP.hpp.
◆ pep_alphabet
| const char* Sequence::pep_alphabet = "[^ARNDBCQEZGHILKMFPSTWYV\\-]" |
A regex for the complement of an amino acid alphabet
Definition at line 50 of file SeqRegexes.hpp.
◆ SEQMAXDOUBLE
| const double Sequence::SEQMAXDOUBLE = std::numeric_limits<double>::max() |
The maximum value of an double
Definition at line 36 of file SeqConstants.cc.
◆ SEQMAXUNSIGNED
| const unsigned Sequence::SEQMAXUNSIGNED = std::numeric_limits<unsigned>::max() |
The maximum value of an unsinged integer.
Definition at line 32 of file SeqConstants.cc.
