Summary statistics and other analysis of Sequence::VariantMatrixSee Tutorial/overview. More...
Namespaces | |
| Sequence::Recombination | |
| Methods dealing with recombination. | |
Classes | |
| struct | Sequence::PairwiseLDstats |
| Pairwise linkage disequilibrium (LD) stats . More... | |
| struct | Sequence::AlleleCounts |
| struct | Sequence::GarudStats |
| struct | Sequence::nSLiHS |
| class | Sequence::PolySIM |
| Analysis of coalescent simulation data. More... | |
| class | Sequence::PolySNP |
| Molecular population genetic analysis. More... | |
| class | Sequence::PolyTableSlice< T > |
| A container class for "sliding windows" along a polymorphism table. More... | |
| class | Sequence::FST |
analysis of population structure using 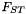 More... More... | |
Functions | |
| std::pair< double, double > | Sequence::nSL (const std::size_t &core, const SimData &d, const std::unordered_map< double, double > &gmap=std::unordered_map< double, double >()) __attribute__((deprecated)) |
| template<typename F > | |
| void | Sequence::sstats_algo::aggregate_sites (const VariantMatrix &m, const F &f, const std::int8_t refstate) |
| template<typename F > | |
| void | Sequence::sstats_algo::aggregate_sites (const VariantMatrix &m, const F &f, const std::vector< std::int8_t > &refstates) |
| std::vector< AlleleCounts > | Sequence::allele_counts (const AlleleCountMatrix &m) |
| Count number of alleles at each site. More... | |
| std::vector< AlleleCounts > | Sequence::non_reference_allele_counts (const AlleleCountMatrix &m, const std::int8_t refstate) |
| Count number of non-reference alleles at each site. More... | |
| std::vector< AlleleCounts > | Sequence::non_reference_allele_counts (const AlleleCountMatrix &m, const std::vector< std::int8_t > &refstates) |
| Count number of non-reference alleles at each site. More... | |
| double | Sequence::tajd (const AlleleCountMatrix &ac) |
| Tajima's D. More... | |
| double | Sequence::hprime (const AlleleCountMatrix &ac, const std::int8_t refstate) |
| double | Sequence::hprime (const AlleleCountMatrix &m, const std::vector< std::int8_t > &refstates) |
| double | Sequence::faywuh (const AlleleCountMatrix &ac, const std::int8_t refstate) |
| Fay and Wu's H. More... | |
| double | Sequence::faywuh (const AlleleCountMatrix &ac, const std::vector< std::int8_t > &refstates) |
| Fay and Wu's H. More... | |
| std::vector< std::int32_t > | Sequence::difference_matrix (const VariantMatrix &m) |
| Calculate number of differences between all samples. More... | |
| std::vector< std::int32_t > | Sequence::label_haplotypes (const VariantMatrix &m) |
| Assign a unique label to each haplotype. More... | |
| std::int32_t | Sequence::number_of_haplotypes (const VariantMatrix &m) |
| Calculate the number of haplotypes in a sample. More... | |
| double | Sequence::haplotype_diversity (const VariantMatrix &m) |
| Calculate the haplotype diversity of a sample. More... | |
| std::int32_t | Sequence::rmin (const VariantMatrix &m) |
| std::uint32_t | Sequence::nvariable_sites (const AlleleCountMatrix &m) |
| Number of polymorphic sites. More... | |
| std::uint32_t | Sequence::nbiallelic_sites (const AlleleCountMatrix &m) |
| Number of bi-allelic sites. More... | |
| std::uint32_t | Sequence::total_number_of_mutations (const AlleleCountMatrix &m) |
| Total number of mutations in the sample. More... | |
| double | Sequence::thetah (const AlleleCountMatrix &ac, const std::int8_t refstate) |
Fay and Wu's 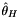 . More... . More... | |
| double | Sequence::thetah (const AlleleCountMatrix &m, const std::vector< std::int8_t > &refstates) |
| Fay and Wu's . More... | |
| double | Sequence::thetal (const AlleleCountMatrix &ac, const std::int8_t refstate) |
Zeng et al. 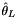 . More... . More... | |
| double | Sequence::thetal (const AlleleCountMatrix &m, const std::vector< std::int8_t > &refstates) |
| Zeng et al. . More... | |
| double | Sequence::thetapi (const AlleleCountMatrix &ac) |
| Mean pairwise differences. More... | |
| double | Sequence::thetaw (const AlleleCountMatrix &ac) |
| Watterson's theta. More... | |
| nSLiHS | Sequence::nsl (const VariantMatrix &m, const std::size_t core, const std::int8_t refstate) |
| nSL and iHS statistics More... | |
Detailed Description
Summary statistics and other analysis of Sequence::VariantMatrix
See Tutorial/overview.
Function Documentation
◆ aggregate_sites() [1/2]
|
inline |
Helper algorithm for implementing summary statistics.
Several common summary statistics are combinations of others. Examples include Tajima's D, Fay and Wu's H, etc.. If we take D as an example, it is tempting to use existing functions, such as Sequence::thetapi and Sequence::thetaw, as intermediate steps. However, doing so goes over the data multiple times.
Fortunately, these statistics are often easy enough to implement that we could calculate pi and Watterson's theta in one loop. This function helps you do that.
- Parameters
-
m A VariantMatrix f A function taking a const StateCounts & and returning nothing. refstate The reference state.
This function loops over m.nsites and passes the state counts on to the aggregator function f.
See the implementation of Sequence::tajd for an example.
Definition at line 18 of file algorithm.hpp.
◆ aggregate_sites() [2/2]
|
inline |
Helper algorithm for implementing summary statistics.
Several common summary statistics are combinations of others. Examples include Tajima's D, Fay and Wu's H, etc.. If we take D as an example, it is tempting to use existing functions, such as Sequence::thetapi and Sequence::thetaw, as intermediate steps. However, doing so goes over the data multiple times.
Fortunately, these statistics are often easy enough to implement that we could calculate pi and Watterson's theta in one loop. This function helps you do that.
- Parameters
-
m A VariantMatrix f A function taking a const StateCounts & and returning nothing. refstates Vector of reference states
This function loops over m.nsites and passes the state counts on to the aggregator function f.
See the implementation of Sequence::hprime for an example.
Definition at line 56 of file algorithm.hpp.
◆ allele_counts()
| count_type Sequence::allele_counts | ( | const AlleleCountMatrix & | m | ) |
Count number of alleles at each site.
- Parameters
-
m An AlleleCountMatrix
Definition at line 50 of file allele_counts.cc.
◆ difference_matrix()
| std::vector< std::int32_t > Sequence::difference_matrix | ( | const VariantMatrix & | m | ) |
Calculate number of differences between all samples.
- Parameters
-
m A VariantMatrix
- Returns
- std::vector<std::int32_t>
For 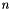 samples in m, the output contains
samples in m, the output contains 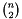 elements. More concretely, the elements are populated according to:
elements. More concretely, the elements are populated according to:
Missing data to not contribute to differences between sequences. Thus, low-quality data may lead to uninformative return values.
Included via Sequence/summstats.hpp or Sequence/summstats/classics.hpp
Definition at line 15 of file haplotype_statistics.cc.
◆ faywuh() [1/2]
| double Sequence::faywuh | ( | const AlleleCountMatrix & | ac, |
| const std::int8_t | refstate | ||
| ) |
Fay and Wu's H.
- Parameters
-
m An AlleleCountMatrix refstate The ancestral state.
- Returns
- Fay and Wu's H, or nan if m is empty/invariant.
- Note
- It is an error to consider refstate as anything other than the ancestral state for each site.
This function is included via Sequence/summstats.hpp, Sequence/summstats/classics.hpp Sequence/summstats/thetah.hpp
See [2] for details.
◆ faywuh() [2/2]
| double Sequence::faywuh | ( | const AlleleCountMatrix & | ac, |
| const std::vector< std::int8_t > & | refstates | ||
| ) |
Fay and Wu's H.
- Parameters
-
m An AlleleCountMatrix refstates The ancestral state at each site.
- Returns
- Fay and Wu's H, or nan if m is empty/invariant.
- Note
- It is an error to consider refstates as anything other than the ancestral state at each site.
This function is included via Sequence/summstats.hpp, or Sequence/summstats/classics.hpp
See [2] for details.
◆ haplotype_diversity()
| double Sequence::haplotype_diversity | ( | const VariantMatrix & | m | ) |
Calculate the haplotype diversity of a sample.
- Parameters
-
m A VariantMatrix
- Returns
- Haplotype heterozygosity, a double.
The "haplotype heterozygosity" is calculated by counting haplotype labels (see label_haplotypes).
- Note
- The value nan is returned if m.nsam == 0. If m contains data, but all sites are invariant, the function returns 0.0. See testClassicSummstatsEmptyVariantMatrix.cc for examples.
Included via Sequence/summstats.hpp or Sequence/summstats/classics.hpp
See [1] for details.
Definition at line 134 of file haplotype_statistics.cc.
◆ hprime() [1/2]
| double Sequence::hprime | ( | const AlleleCountMatrix & | ac, |
| const std::int8_t | refstate | ||
| ) |
The H' statistic
- Parameters
-
m An AlleleCountMatrix refstate How the ancestral state is encoded.
- Returns
- H'
- Note
- nan is returned if m is empty/invariant. It is an error to consider refstate as anything other than the ancestral state.
See [10] for details.
Included via Sequence/summstats.hpp or Sequence/summstats/classics.hpp
◆ hprime() [2/2]
| double Sequence::hprime | ( | const AlleleCountMatrix & | m, |
| const std::vector< std::int8_t > & | refstates | ||
| ) |
The H' statistic
- Parameters
-
m An AlleleCountMatrix refstates A vector of ancestral states, equal in length to m.sites
- Returns
- H'
- Note
- nan is returned if m is empty/invariant. It is an error to consider refstates as anything other than the ancestral state at each site.
See [10] for details.
Included via Sequence/summstats.hpp or Sequence/summstats/classics.hpp
◆ label_haplotypes()
| std::vector< std::int32_t > Sequence::label_haplotypes | ( | const VariantMatrix & | m | ) |
Assign a unique label to each haplotype.
- Parameters
-
m A VariantMatrix
- Returns
- std::vector<std::int32_t>
If there are 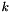 unique samples in m, which represents a sample of size nsam, the return value contains nsam elements whose values are
unique samples in m, which represents a sample of size nsam, the return value contains nsam elements whose values are 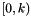 for "good" input. Here, "bad" input means that some of the samples consist entirely of missing data. In that case, they are given the label of -1.
for "good" input. Here, "bad" input means that some of the samples consist entirely of missing data. In that case, they are given the label of -1.
This function is implemented via a call to difference_matrix.
Included via Sequence/summstats.hpp or Sequence/summstats/classics.hpp
Definition at line 75 of file haplotype_statistics.cc.
◆ nbiallelic_sites()
| std::uint32_t Sequence::nbiallelic_sites | ( | const AlleleCountMatrix & | m | ) |
Number of bi-allelic sites.
Return the number of sites with exactly two non-missing states.
- Parameters
-
m An AlleleCountMatrix
- Returns
- std::uint32_t
Definition at line 28 of file nvariablesites.cc.
◆ non_reference_allele_counts() [1/2]
| count_type Sequence::non_reference_allele_counts | ( | const AlleleCountMatrix & | m, |
| const std::int8_t | refstate | ||
| ) |
Count number of non-reference alleles at each site.
- Parameters
-
m An AlleleCountMatrix m refstate The reference state for all sites.
Definition at line 80 of file allele_counts.cc.
◆ non_reference_allele_counts() [2/2]
| count_type Sequence::non_reference_allele_counts | ( | const AlleleCountMatrix & | m, |
| const std::vector< std::int8_t > & | refstates | ||
| ) |
Count number of non-reference alleles at each site.
- Parameters
-
m An AlleleCountMatrix m refstate The reference state at each site.
Definition at line 62 of file allele_counts.cc.
◆ nsl()
| nSLiHS Sequence::nsl | ( | const VariantMatrix & | m, |
| const std::size_t | core, | ||
| const std::int8_t | refstate | ||
| ) |
nSL and iHS statistics
- Parameters
-
m A VariantMatrix core The index of the core site refstate The value of the reference/ancestral allelic state
- Returns
- an nSLiHS object
See nSL_from_ms.cc for example
See [3] for details.
- Examples:
- nSL_from_ms.cc.
◆ nSL()
| pair< double, double > Sequence::nSL | ( | const std::size_t & | core, |
| const SimData & | d, | ||
| const std::unordered_map< double, double > & | gmap = std::unordered_map<double, double>() |
||
| ) |
The nSL statistic of Ferrer-Admetlla et al. doi: 10.1093/molbev/msu077.
- Parameters
-
core The index of the "focal/core" SNP d An object of type Sequence::SimData gmap The positions of every marker in d on the genetic map. If std::unordered_map<double,double>() is passed, iHS is calculated using SNP positions.
- Returns
- nSL and iHs, with the latter as defined in doi: 10.1093/molbev/msu077.
- Note
- This routine was validated by comparing to code provided by Ferrer-Admetlla et al.
- Warning
- The use of 'gmap' is untested.
◆ number_of_haplotypes()
| std::int32_t Sequence::number_of_haplotypes | ( | const VariantMatrix & | m | ) |
Calculate the number of haplotypes in a sample.
- Parameters
-
m A VariantMatrix
- Returns
- The number of unique elements in the sample, std::int32_t
This returns the number of unique columns in m.
- Note
- The value -1 is returned if m.nsam == 0. If m contains data, but all sites are invariant, the function returns 1. See testClassicSummstatsEmptyVariantMatrix.cc for examples.
Include via Sequence/summstats.hpp or Sequence/summstats/classics.hpp
See [1] for details.
Definition at line 117 of file haplotype_statistics.cc.
◆ nvariable_sites()
| std::uint32_t Sequence::nvariable_sites | ( | const AlleleCountMatrix & | m | ) |
Number of polymorphic sites.
Returns the number of sites with more than one non-missing state
- Parameters
-
m An AlleleCountMatrix
- Returns
- std::uint32_t
Definition at line 8 of file nvariablesites.cc.
◆ rmin()
| std::int32_t Sequence::rmin | ( | const VariantMatrix & | m | ) |
Hudson and Kaplan's Rmin statistic
- Parameters
-
m A VariantMatrix
- Returns
- Rmin, std::int32_t
Included via Sequence/summstats.hpp or Sequence/summstats/classics.hpp
See [5] for details.
◆ tajd()
| double Sequence::tajd | ( | const AlleleCountMatrix & | ac | ) |
Tajima's D.
- Parameters
-
m An AlleleCountMatrix
- Returns
- Tajima's D
- Note
- nan is returned if m is empty/invariant.
See [7] for details.
Included via Sequence/summstats.hpp or Sequence/summstats/classics.hpp
◆ thetah() [1/2]
| double Sequence::thetah | ( | const AlleleCountMatrix & | ac, |
| const std::int8_t | refstate | ||
| ) |
Fay and Wu's .
- Parameters
-
m An AlleleCountMatrix refstate The ancestral state
- Returns
- double
See [2] for details.
Definition at line 117 of file thetah_thetal.cc.
◆ thetah() [2/2]
| double Sequence::thetah | ( | const AlleleCountMatrix & | m, |
| const std::vector< std::int8_t > & | refstates | ||
| ) |
Fay and Wu's .
- Parameters
-
m a VariantMatrix refstate Vector of ancestral states.
- Returns
- double
See [2] for details.
Definition at line 123 of file thetah_thetal.cc.
◆ thetal() [1/2]
| double Sequence::thetal | ( | const AlleleCountMatrix & | ac, |
| const std::int8_t | refstate | ||
| ) |
Zeng et al. .
- Parameters
-
m An AlleleCountMatrix refstate The ancestral state
- Returns
- double
See [10] for details.
Definition at line 130 of file thetah_thetal.cc.
◆ thetal() [2/2]
| double Sequence::thetal | ( | const AlleleCountMatrix & | m, |
| const std::vector< std::int8_t > & | refstates | ||
| ) |
Zeng et al. .
- Parameters
-
m An AlleleCountMatrix refstate Vector of ancestral states.
- Returns
- double
See [10] for details.
Definition at line 136 of file thetah_thetal.cc.
◆ thetapi()
| double Sequence::thetapi | ( | const AlleleCountMatrix & | ac | ) |
Mean pairwise differences.
- Parameters
-
m An AlleleCountMatrix
- Returns
- Mean pairwise differences
- Note
- Calcuated as sum over one minus site homozygosity
This function is included via Sequence/summstats.hpp, Sequence/summstats/classics.hpp or Sequence/summstats/thetapi.hpp
See [6] for details.
Definition at line 7 of file thetapi.cc.
◆ thetaw()
| double Sequence::thetaw | ( | const AlleleCountMatrix & | ac | ) |
Watterson's theta.
- Parameters
-
m An AlleleCountMatrix
- Returns
- Watterson's theta, a double
- Note
- For a site with states,
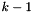 is added to the number of inferred mutations. In other words, the calculation is based on the total number of mutations.
is added to the number of inferred mutations. In other words, the calculation is based on the total number of mutations.
See [9] for details.
◆ total_number_of_mutations()
| std::uint32_t Sequence::total_number_of_mutations | ( | const AlleleCountMatrix & | m | ) |
Total number of mutations in the sample.
Return 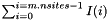 where
where 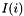 is
is 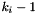 if
if 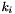 , the number of states at the
, the number of states at the 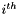 site, is greater than one, and zero otherwise.
site, is greater than one, and zero otherwise.
- Parameters
-
m An AlleleCountMatrix
- Returns
- std::uint32_t
Definition at line 48 of file nvariablesites.cc.
