Class Hierarchy
This inheritance list is sorted roughly, but not completely, alphabetically:
[detail level 12]
| CSequence::_PolySNPImpl | |
| ▼CSequence::AlignStream< T > | Virtual interface to alignment streams |
| CSequence::ClustalW< T > | ClustalW streams |
| CSequence::phylipData< T > | |
| CSequence::AlleleCountMatrix | Matrix representation of allele counts in a VariantMatrix To be constructed |
| CSequence::AlleleCounts | |
| CSequence::ambiguousNucleotide | Tests if a character is in the set A,G,C,T |
| CSequence::coalsim::chromosome | A chromosome is a container of segments |
| CSequence::internal::col_view_< T > | Implementation details for Sequence::ColView and Sequence::ConstColView |
| CSequence::col_view_iterator< POINTER > | Iterator for column views |
| CSequence::Comeron95 | Ka and Ks by Comeron's (1995) method |
| CSequence::ComplementBase | |
| CSequence::countDerivedStates | Functor to count the number of derived states, excluding gaps and missing data, in a range of characters |
| Cfasta_constructors_fixture | \ file FastaConstructors.cc |
| Cfirst_is_equal< key, value > | Functor that checks for equality of first member of two pairs |
| CSequence::FST | Analysis of population structure using 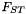 |
| CSequence::GarudStats | |
| CSequence::Grantham | Grantham's distances |
| CSequence::HKAdata | Data from a single locus for an HKA test |
| CSequence::HKAresults | Results of calculations of the HKA test |
| CSequence::invalidPolyChar | This functor can be used to determine if a range contains characters that the SNP analysis routines in this library cannot handle gracefully |
| CSequence::Kimura80 | Kimura's 2-parameter distance |
| CSequence::coalsim::marginal | The genealogy of a portion of a chromosome on which no recombination has occurred |
| CSequence::coalsim::newick_stream_marginal_tree | Class that provides a typecast-on-output of a marginal tree to a newick tree Example use: |
| CSequence::coalsim::newick_stream_marginal_tree_impl | |
| CSequence::nmuts< counter > | Calculate the number of mutations at a polymorphic site |
| CSequence::coalsim::node | A point on a marginal tree at which a coalescent event occurs |
| CSequence::nSLiHS | |
| CoutputFreq | |
| CSequence::PairwiseLDstats | Pairwise linkage disequilibrium (LD) stats |
| CSequence::PolySIM | Analysis of coalescent simulation data |
| CSequence::PolySNP | Molecular population genetic analysis |
| ▼CSequence::PolyTable | The base class for polymorphism tables |
| CSequence::PolySites | Polymorphism tables for sequence data |
| CSequence::PolyTableSlice< T > | A container class for "sliding windows" along a polymorphism table |
| CSequence::RedundancyCom95 | Calculate redundancy of a genetic code using Comeron's counting scheme |
| CSequence::internal::row_view_< T > | Implementation details for Sequence::RowView and Sequence::ConstRowView |
| CSequence::coalsim::segment | A portion of a recombining chromosome |
| ▼CSequence::Seq | Abstract interface to sequence objects |
| CSequence::Fasta | FASTA sequence stream |
| CSequence::fastq | |
| CSequence::shortestPath | Calculate shortest path between 2 codons |
| CSequence::SimData | Data from coalescent simulations |
| CSequence::SimParams | Parameters for Hudson's simulation program |
| CSequence::SimpleSNP | SNP table data format |
| CSequence::SingleSub | Deal with codons differing at 1 position |
| CSequence::Sites | Calculate length statistics for divergence calculations |
| Cssh | Calculate nucleotide diversity from a polymorphic site |
| CSequence::stateCounter | Keep track of state counts at a site in an alignment or along a sequence |
| CSequence::StateCounts | Track character state occurrence at a site in a VariantMatrix |
| CSequence::Sums< T > | |
| CSequence::ThreeSubs | Deal with codons differing at all 3 positions |
| CSequence::TwoSubs | Deal with codons differing at 2 positions |
| CSequence::VariantMatrix | Matrix representation of variation data |
| ▼CSequence::WeightingScheme2 | Abstract interface to weighting schemes when codons differ at 2 positions |
| CSequence::GranthamWeights2 | Weights paths by Grantham's distances for codons differing at 2 sites |
| CSequence::Unweighted2 | Weights all pathways equally |
| ▼CSequence::WeightingScheme3 | Abstract interface to weighting schemes when codons differ at 3 positions |
| CSequence::GranthamWeights3 | Weights paths by Grantham's distances for codons differing at 3 sites |
| CSequence::Unweighted3 | Weights all pathways equally |
