Adaptation of a trait to different optima in different demes#
Show code cell source
import fwdpy11
import demes
import demesdraw
import numpy as np
First, set up some basic parameters that we will need:
# diploid population size:
N=1000
genome_length=1e8
We will simulate a simple model of a population split:
yaml=f"""
time_units: generations
demes:
- name: ancestor
epochs:
- end_time: 100
start_size: {N}
- name: deme0
ancestors: [ancestor]
epochs:
- start_size: {N}
- name: deme1
ancestors: [ancestor]
epochs:
- start_size: {N}
"""
demesdraw.tubes(demes.loads(yaml));
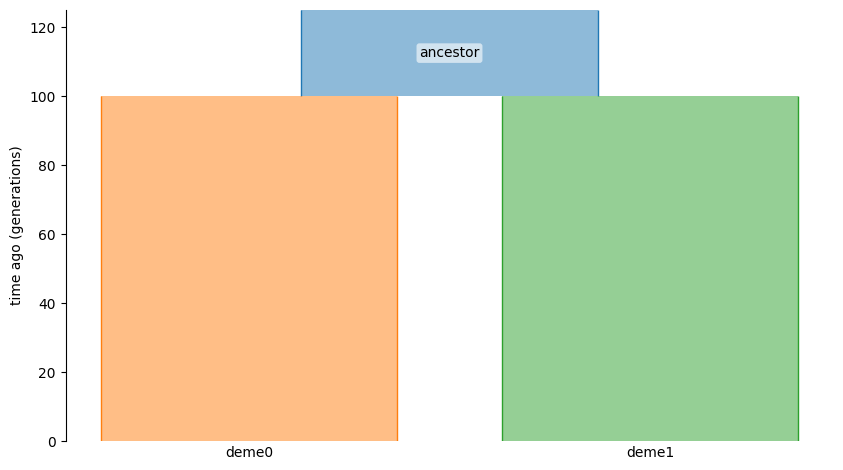
There are three demes in this model: the ancestor and the two descendants. We want to have each descendant deme evolve to a different phenotypic optimum. To do this, we must supply a genetic value calculation for each of the three demes in the model:
optima_ancestor = [fwdpy11.Optimum(when=0, VS=1.0, optimum=0.0)]
optima_deme0 = [fwdpy11.Optimum(when=0, VS=1.0, optimum=0.0),
fwdpy11.Optimum(when=10*N, VS=1.0, optimum=1.0)
]
optima_deme1 = [fwdpy11.Optimum(when=0, VS=1.0, optimum=0.0),
fwdpy11.Optimum(when=10*N, VS=1.0, optimum=-1.0)
]
gss_ancestor = fwdpy11.GaussianStabilizingSelection.single_trait(optima=optima_ancestor)
gss0 = fwdpy11.GaussianStabilizingSelection.single_trait(optima=optima_deme0)
gss1 = fwdpy11.GaussianStabilizingSelection.single_trait(optima=optima_deme1)
We will model additive effects on the trait:
gvalue = [fwdpy11.Additive(scaling=2.0, gvalue_to_fitness=gss_ancestor),
fwdpy11.Additive(scaling=2.0, gvalue_to_fitness=gss0),
fwdpy11.Additive(scaling=2.0, gvalue_to_fitness=gss1)]
We will model effect sizes as constant across demes.
We can use fwdpy11.GaussianS to model a Gaussian distribution of effect sizes:
dfe = fwdpy11.GaussianS(0, genome_length, 1.0, sd=0.1)
The rest of the setup is straightforward. We will evolve the population and print out the final mean trait values, which will be very close to the optima of each descendant deme.
Show code cell source
demog = fwdpy11.ForwardDemesGraph.from_demes(yaml, burnin=10)
pdict = {
'sregions': [dfe],
'recregions': [fwdpy11.BinomialInterval(0, genome_length, 0.5)],
'rates': (0, 5e-3, None),
'demography': demog,
'gvalue': gvalue,
'simlen': 10*N + 100,
'prune_selected': False,
}
params = fwdpy11.ModelParams(**pdict)
pop = fwdpy11.DiploidPopulation(N, genome_length)
rng = fwdpy11.GSLrng(54321)
fwdpy11.evolvets(rng, pop, params, simplification_interval=100)
md = np.array(pop.diploid_metadata)
for deme in np.unique(md["deme"]):
w = np.where(md["deme"] == deme)
print(f"mean trait value in deme {deme} = {md["g"][w].mean()}")
mean trait value in deme 1 = 0.9288320707795822
mean trait value in deme 2 = -1.0138504399491264
To see how to extend this model to allow the effect sizes to differ across demes, see here.