Mutations with variable dominance#
Added in version 0.13.0.
The heterozygous effect of a mutation does not need to be constant.
You may use instances of classes derived from fwdpy11.MutationDominance to assign functions to generate the dominance of mutations.
The available classes are:
fwdpy11.FixedDominanceThis class is equivalent to passing in afloatto thehkwarg. See here
Example#
import fwdpy11
des = fwdpy11.GaussianS(beg=0, end=1, weight=1, sd=0.1,
h=fwdpy11.LargeEffectExponentiallyRecessive(k=5.0))
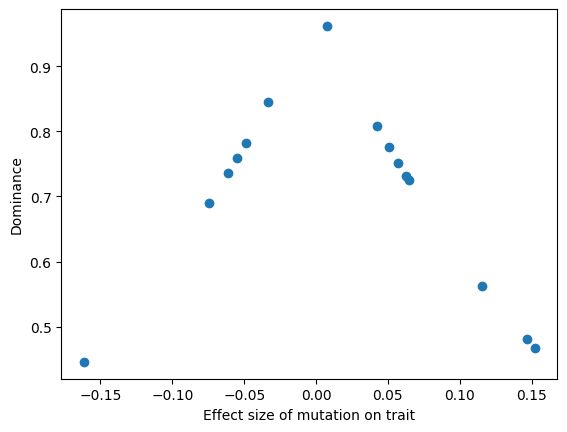
If we apply this des object to a model of a quantitative trait evolving to a sudden “optimum shift”, then we see that the larger effect variants present at the end of the simulation to indeed have smaller dominance coefficients.
Show code cell source
pop = fwdpy11.DiploidPopulation(500, 1.0)
rng = fwdpy11.GSLrng(54321)
optima = [
fwdpy11.Optimum(when=0, optimum=0.0, VS=1.0),
fwdpy11.Optimum(when=10 * pop.N - 200, optimum=1.0, VS=1.0),
]
GSS = fwdpy11.GaussianStabilizingSelection.single_trait(optima)
rho = 1000.
p = {
"nregions": [],
"gvalue": fwdpy11.Additive(2.0, GSS),
"sregions": [des],
"recregions": [fwdpy11.PoissonInterval(0, 1., rho / float(4 * pop.N))],
"rates": (0.0, 1e-3, None),
"prune_selected": False,
"demography": fwdpy11.ForwardDemesGraph.tubes([pop.N], burnin=10),
"simlen": 10 * pop.N,
}
params = fwdpy11.ModelParams(**p)
fwdpy11.evolvets(rng, pop, params, 100, suppress_table_indexing=True)
Show code cell source
import matplotlib.pyplot as plt
esize = [pop.mutations[m.key].s for m in pop.tables.mutations]
h = [pop.mutations[m.key].h for m in pop.tables.mutations]
f, ax = plt.subplots()
ax.scatter(esize, h)
ax.set_xlabel("Effect size of mutation on trait")
ax.set_ylabel("Dominance")
plt.show();
Using discrete distributions#
fwdpy11.DiscreteDESD specifies a Discrete Effect Size and Dominance joint distribution.
A list of tuple of (effect size, dominance, weight) specify the joint distribution.
For example:
import math
import numpy as np
joint_dist = []
for s in np.arange(0.1, 1, 0.1):
joint_dist.append((-s, math.exp(-s), 1./s))
des = fwdpy11.DiscreteDESD(beg=0, end=1, weight=1, joint_dist=joint_dist)
print(des)
fwdpy11.DiscreteDESD(beg=0, end=1, weight=1, joint_dist=[(np.float64(-0.1), 0.9048374180359595, np.float64(10.0)), (np.float64(-0.2), 0.8187307530779818, np.float64(5.0)), (np.float64(-0.30000000000000004), 0.7408182206817179, np.float64(3.333333333333333)), (np.float64(-0.4), 0.6703200460356393, np.float64(2.5)), (np.float64(-0.5), 0.6065306597126334, np.float64(2.0)), (np.float64(-0.6), 0.5488116360940264, np.float64(1.6666666666666667)), (np.float64(-0.7000000000000001), 0.49658530379140947, np.float64(1.4285714285714284)), (np.float64(-0.8), 0.44932896411722156, np.float64(1.25)), (np.float64(-0.9), 0.4065696597405991, np.float64(1.1111111111111112))], coupled=True, label=0, scaling=1.0)
The result is that mutations with smaller effect sizes are more common (larger weights) and more dominant.